

引言
在过去的几年中,数据非依赖采集 (DIA) 方法在蛋白质组鉴定与定量分析流程中日益成为标准。随着DIA技术的广泛应用,各种数据处理工具应运而生,其性能和灵敏度均稳步提升。然而,挑战依然存在——例如,如何处理共洗脱的碎片离子、应对海量数据,以及解决因DIA数据的组合特性而导致的计算复杂性。
Protein Metrics, LLC的MS/MS搜索引擎在数据依赖采集 (DDA) 应用于翻译后修饰 (PTM) 工作流程中享有盛誉,已被研究人员和制药公司广泛采用。基于这一基础,Protein Metrics最近推出了一款专为支持DIA数据而设计的新工作流程。本文介绍了为DIA分析量身定制的高性能、可扩展算法原型的开发,这标志着我们在处理复杂蛋白质组数据集方面的能力取得了重大进展。
方法
这款新型搜索引擎的原型在现有的Byos®宿主细胞蛋白workflow (HCP)和多蛋白定量(MPQ) module流程框架内进行了评估。在这些测试中,新型引擎被用于替代Protein Metrics传统的Byonic™搜索引擎。
结果
比较肽段和蛋白质异构体的检测/定量能力
在本研究中,我们通过引入一款专为适配DIA数据特性而设计的新型搜索引擎,探索了扩展Byos®平台DIA分析能力的新方法。我们的工作聚焦于提升以下三个关键方面:
1、通过利用多同位素信息,增强对离子信号的灵敏度,同时保持对可能影响单个同位素的潜在干扰的稳定性。
2、优化打分系统,以便更好地区分真实匹配和干扰信号。
3、更精准地检测前体和碎片离子信号的共洗脱。
我们的方法围绕在整个分析流程中保持灵活性展开:
-
在最终评分阶段之前,我们会保留所有在多种电荷态和不同假设条件下匹配到的碎片信号。这确保了任何可能具有价值的匹配都不会被过早舍弃。
-
新的搜索引擎支持将所有相关的同位素峰(无论是来自棒状还是轮廓模式数据)与观测到的信号进行匹配。
-
通过纳入轮廓数据,我们提高了对真实信号的辨别能力,即使在存在复杂同位素干扰的情况下,也能实现匹配。尤为独特的是,这种方法允许对原始轮廓数据点进行直接匹配。我们利用多个同位素观测结果来优化前体离子和碎片离子的共洗脱。
-
最后,我们汇总前体离子峰顶附近多个相邻MS2扫描的信号,以进一步提高匹配的辨别能力和边界精度。
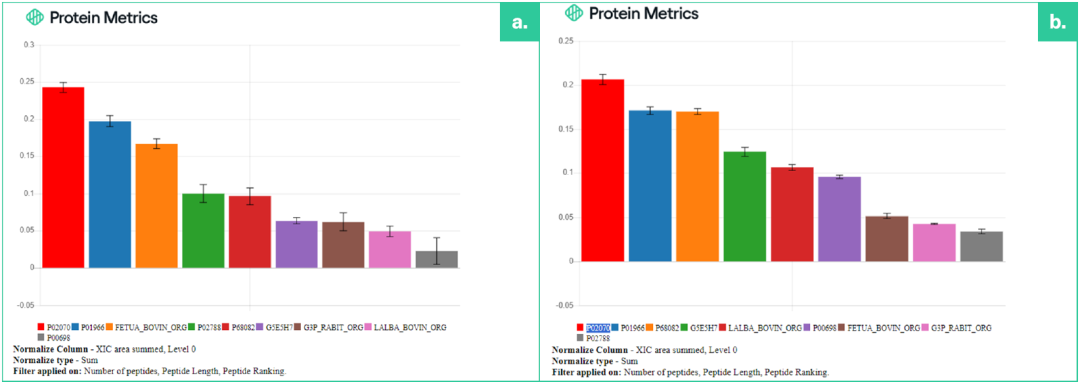
图1. 现有的采用Byonic™的Byos®MPQ工作流程(a) 和配备原型搜索引擎的新型Byos® MPQ DIA工作流程 (b) 均成功量化了所有九种添加的蛋白质。然而,这两种方法在重复运行中表现出的量化变异性水平不同。
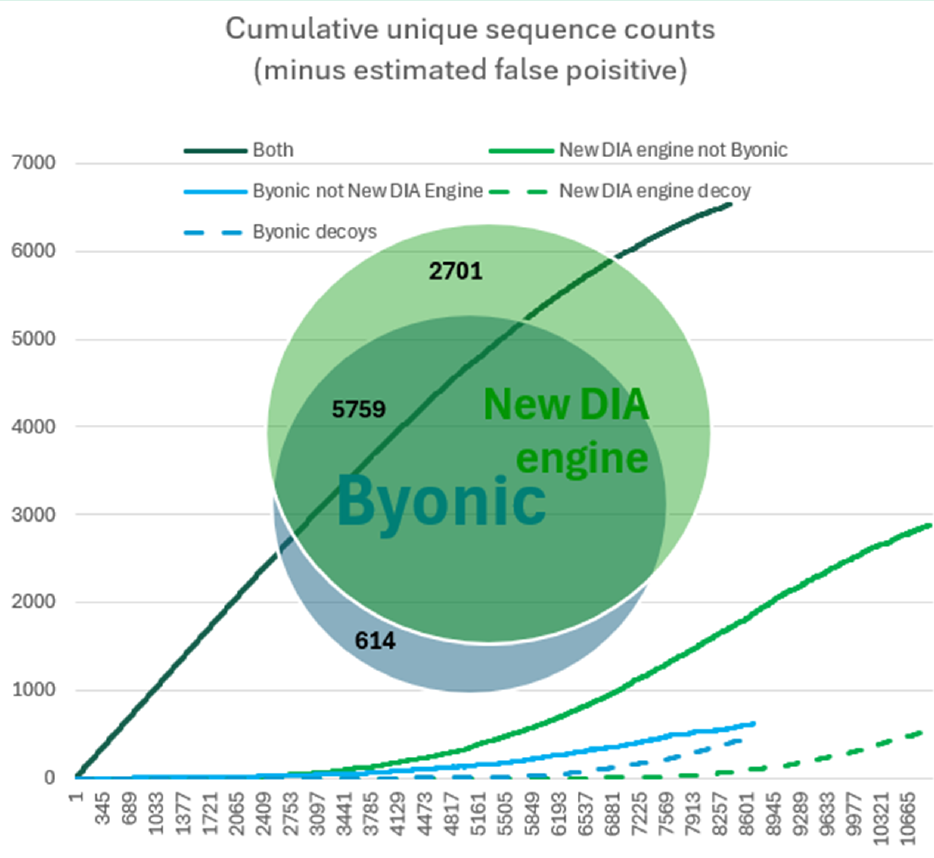
图2. 两款引擎间独特序列鉴定结果的比较。这些图以10%的FDR阈值为限制条件,而韦恩图中的数字是基于1%的FDR计算得出的。
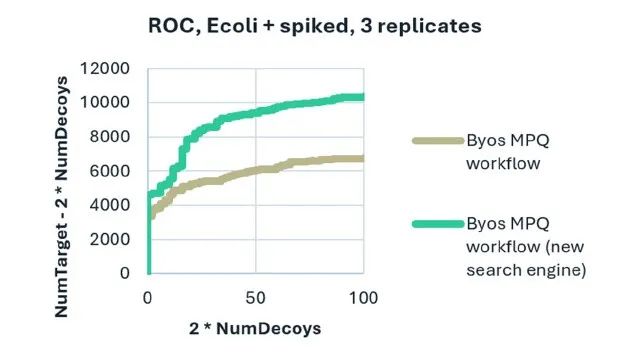
图3. 通过反向数据库搜索分析展示出了更高的鉴定率

图4. (a) 由于在远离洗脱峰顶处采集的MS2谱图中噪声增加,Byonic™可能会产生额外的、有时相互矛盾的修饰位点定位结果。(b) 新型搜索引擎整合了洗脱峰顶周围多个扫描的MS2数据,从而提高了修饰位点定位的准确性。
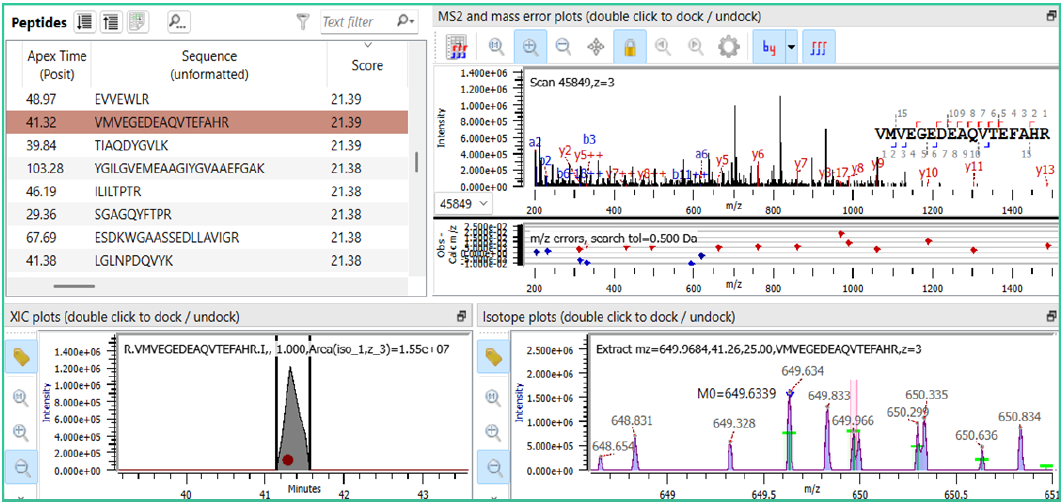
图5. 复杂同位素混合物中鉴定结果的示例
结论
与Byonic™相比,这款新型搜索引擎展现出了对真正阳性匹配结果更强的辨别能力。在涉及复杂同位素干扰的具有挑战性的情况下,它能成功恢复额外的鉴定结果。此外,我们还观察到在DIA数据分析中,其量化精度有所提高,这体现在变异系数 (CV) 值降低上。而且,新方法显示出偶发性异构体报告频率的降低,这表明鉴定结果具有更高的一致性和可靠性。
作者
Ignat Shilov, Christian Heckendorf, Abhishek Roushan, Gary Wilson, Claire Bramwell
Protein Metrics, Boston MA
推荐阅读
END