


New Year’s Greetings from Protein Metrics
福鼠呈祥 恭喜发财 万事如意
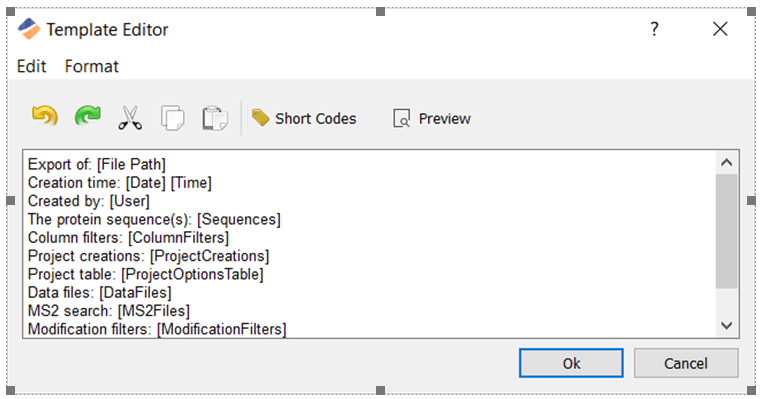
本期Protein Metrics Insights,我们将介绍: 1) 利用Byos基本和高级报告功能灵活定制报告; 2) 利用SV auto-comment功能自动验证。 1. 报告功能 Byos系统默认的分析流程(Workflow)已经配置了该Workflow下适合用户的报告。用户可以在此基础上根据自身需求和其客户的要求而进行定制化报告并将定制化后的报告存为报告模板,以便重复应用在以后的项目中。 日常分析中在Byos中创建项目后,报告会自动生成,并且在Byos界面中显示成一个标签页(Tab)。报告的形式可以根据实际需求进行定制。 一个完整的报告分成多个标签页(Tab)。第一个Tab为分析结果的总结(Summary)。在Peptide Workflow中,默认的Summary页中包含蛋白覆盖率(protein coverage)、创建项目时设定的项目选项(project creation options)、用于鉴定的检索参数、 原始数据文件存储位置和输出结果的存储位置等信息。点击菜单栏Edit→Current tab settings,然后点击“Edit summary template”,用户可以对该Summary页进行自定义修改,如删除该视图中的文本和短代码(Short Codes)或对视图的信息进行重新排序(见下图)。 对于高级用户,这个编辑器支持使用基本的html标签(Html Tag)。例如,用户在Summary 页的顶部输入<h2>Study conclusion</h2>,添加Study conclusion为二级标题。 其余页面的数据透视表(Pivot Table)可以用不同的方法进行配置。点击“show configuration”按钮,用户可以个性化地定制在报告中显示哪些内容。 点击“show configuration”后,左边的两列是可定制的部分。最左的一列显示的是允许用户添加到Pivot Table中的可选字段(Field)。第二列显示的是实际在报告中显示的字段。如果需要从最左边选择某个字段添加到表中,点击该字段按钮,然后拖至第二列即可完成。如需将某个当前显示的列从表中移除,只需在第二列中将该字段拖动至最左列。 …
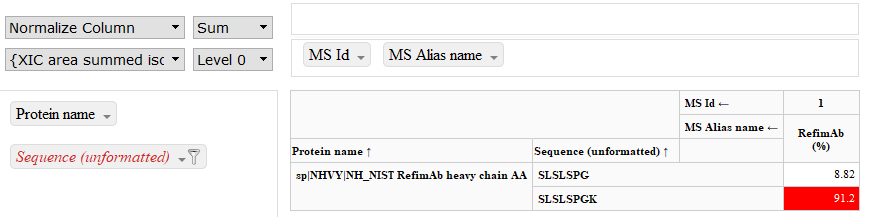
本期Protein Metrics Insights中, 我们将介绍利用Byonic的cleavage flags功能定义蛋白的C-末端赖氨酸剪切(protein C-term Lysine clipping),也将展示升级到最新版本后全新的Intact使用界面。 1. 高级功能 Cleavage Flags 我们经常碰到这样的问题:用户是否需要在Byonic中将蛋白的C-端赖氨酸剪切设定成一种修饰? 是否需要完全取决于用户想如何在报告中显示赖氨酸剪切。 Byonic默认搜索所有蛋白质链N端和C端的单个残基剪切。如果用户在不定义任何修饰的情况下进行全部特定修饰检索(fully-specific search),Byonic会同时鉴定剪切和未剪切的末端肽段。例如,在默认全胰蛋白酶检索中,以下两种结果会同时出现。 K.SLSLSPGK.- K.SLSLSPG.K 它们都会显示在野生型肽段的匹配结果中。用户可以在Byologic的报告中创建一个专门的表格,查看这两种肽段的相对丰度(见下图)。 在上表中,需要注意的是Sequence(unformatted)所在的列添加了筛选功能,因而仅显示选中的两个肽段。用户还可以使用“IsCterm“动态返回列来自动筛选出C-端肽段,无需要手动选择相应的序列。 另一种方法是将赖氨酸酶切设置为”Lys-loss/ -128.094963 @ Protein CTerm K”修饰。采用这种方法时,用户还需要关闭自动搜索单残基剪切的功能。关闭该功能是由Byonic中的“cleavage_flags”命令行来实现。只需在下图中的自定义修饰文本框中输入以下命令: % Custom modification text below cleavage_flags=0 这时赖氨酸缺失(Lys-loss)会被认为是一种修饰,以下两种肽段会被鉴定出来。 K.SLSLSPGK.- K.SLSLSPGK[-128].- 因为赖氨酸缺失是被修饰的肽段,用默认的PTM报告模板生成的报告中会非常直观地显示该肽段的信息(见下图)。 如果用户想更进一步检索C端的甘氨酸缺失和酰胺修饰(Gly-loss+Amide/-58.005479),有以下两种方法可以实现。 1)如果使用cleavage_flags=0命令行,同时Lys-loss设置为“variable-common1”修饰,然后将 Gly-loss+Amide设置为“variable-rare1”修饰,那么系统会同时检索两种修饰。结果报告中的肽段ID为K.SLSLSPG[-58]K[-128].-。 2)如果不使用cleavage_flags=0命令行,设置Gly-loss+Amide为“variable- rare1“修饰,那么结果报告中的肽段ID为K.SLSLSPG[-58].K。 2. 新功能展示 您希望更方便地输入蛋白质的详细信息吗?我们采纳用户的反馈意见,在混合链(combining chains)、二硫键数目的设置(defining the number of disulfide bonds)、Considering clipped species、常见修饰残基的设置、原子量的设定(specifying atomic weights)和生物聚合物类型(DNA, RNA和蛋白)的选择等进行了优化。现在,您可以使用Byos分析寡核苷酸。以下是新版本中完整分子量(Intact)模块里蛋白信息输入的操作界面截图。 …

本期“ Protein Metrics Insights”的重点是Intact Mass。我们将讨论如何对isotopically resolved 谱图进行去卷积分析,然后确定蛋白质的单同位素质量,本期的新功能将介绍Intact Mass的完整分子量重建功能。 同位素分辨的数据去卷积 通常Intact Mass进行去卷积的参数是得到蛋白的平均分子量信息的。如果您的实验室具有同位素分辨的数据并需要去卷积分析,Intact Mass中需要提供特殊的参数,通过参数的设置允许软件在去卷积之后保留原始谱图中的同位素信息。参数的设置也很简单,只需要降低m / z和Mass smoothing 和Spacing等参数的值。以下是一套比较通用的参数: 对于极高分辨率的数据,用户可以进一步降低上述参数以避免数据因Smoothing造成的数据丢失。 Intact Mass进行同位素分辨的数据去卷积分析时,不要忘记在Advance Command中输入如下的命令: [Intact] MaxMonoisotope = 30000 使用此Advance Command后Intact Mass将计算单同位素分子量并将它们放置在Masses表中的“Mass monoisotope”列中。Intact Mass将使用不同的算法去计算所有提取到的峰所对应的单同位素分子量。对于低于30000分子量(MaxMonoisotope参数的值)的信号,Intact Mass将假设质量峰是同位素分辨的,并且将计算的同位素峰与去卷积后峰匹配。对于大于30000的质量,Intact Mass将假设质量峰不是同位素分辨的,它将直接从去卷积的平均分子量中提取校正因子。校正因子是平均分子量质量与平均单一同位素质量之间的差异。 另外一点需要注意的是,MS1彩色点设置为0.6 /电荷内的最高点,这可以防止来自平均质量数的有色点落在同位素分辨的m / z峰之间。 完整分子量重建 分析化学中的一个挑战是如何在系统的研究中能够以正交的方法进行数据结果的对比,从而达到对结论的多维确证。2019年6月发布的Byos/ Intact Mass中,我们引入了一种从肽图谱数据重建蛋白质完整分子量的方法,我们可以通过肽图层面对蛋白酶解之后的肽段和修饰的定量信息,重建理论上的完整分子量信息。在完整蛋白水平上的测得的谱图可以与理论重建的完整分子量谱图进行叠加,通过完整分子量重建的功能,我们提供了一种比较肽图数据与完整分子量数据的方法,这样就可以使用两个谱图之间的差异来比较不同方法之间结果的相关性。 如果您目前正在使用Intact Mass,可以下载beta完整分子量重建演示包可在我们网站的“当前版本”部分中找到。 Upcoming …

Protein Metrics Insights本月起上线了!Insights将为大家介绍一些比较广泛关注的关于软件使用的问题。本期Protein Metrics Insights中,我们将讨论如何对异构化的天冬氨酸进行定量,这也是生物制药表征中的关键质量属性,并介绍了Byos中新上线的游离糖链分析流程。 天冬氨酸异构化的定量 天冬氨酸(Asp,D)和异天冬氨酸(isoAsp,isoD)残基在CID或HCD碎裂模式下是不可区分的, ETD的碎裂模式下,Byonic和Byos可以通过自动识别特征离子z-57和c+57对其进行注释。通常具有D和isoD的肽段在软件中是以同一个Wildtype(未修饰的)肽段的形式显示的,为了计算异构化的相对量,需要将异构的肽段通过保留时间区分并进行定量。 目前在Byos中有手动,半自动和全自动方法来计算每种异构体的相对含量(%PTM),所有这些方法都涉及为含有isoD的肽段创建新肽段并对其进行标记,这样就能够在报告中生成相对百分含量。 下面我们将介绍天然Asp残基的异构化(D→isoD)定量过程,对于Asn脱酰胺形成的异构化的定量(N→D或N→isoD),请参阅我们网站上的应用文献——“PTM Analysis: Asparagine Deamidation Quantified by Byologic”。 半自动化方法 Byos(或Byologic)默认可以检测到多肽XIC中有多个洗脱峰,多个XIC被称为XIC Proposals。XIC Plots视图中可以对XIC Proposals栏进行选择,并得到如下的视图: 操作人员可以在Peptides表中右键单击相应的肽段,然后单击Apply applied XIC splits。这将创建一个新的in-silico肽,并且肽段的XIC自动按照XIC Proposals设置。为了区分D和isoD异构体,将Label添加到新创建的肽中,例如,“isoD”。 全自动方法 当Wildtype肽段显示多个洗脱窗口并且序列中含有预定义的motif(例如DG,DS)时,Byos/Byologic可以自动创建新肽段并将异构体体标记为“isoD”。自动拆分和加入标签需要在项目创建时输入如下Advanced commands 。 [Byologic] AutoSplitPeptides = DG | DS | DD AutoSplitPeptidesLabels …

第67届美国质谱年会将在美国亚特兰大举行,ASMS汇聚了全球质谱分析领域的顶尖科学家和领先的生产厂商,解决方案供应商,是全球质谱领域的一次盛会,Protein Metrics致力于为蛋白质的质谱分析提供更加可靠和便捷的数据分析软件,期待与您在2019 ASMS相遇! 会议期间欢迎您随时莅临Protein Metrics 展台/Booth 416,与我们进行面对面的交流,带您领略蛋白质分析的“小而美”,为您的日常分析提供更好的数据分析解决方案。 Protein Metrics在2019 ASMS中也针对蛋白质数据分析的难点和热点与客户合作发表了部分Poster,小编在此为大家列出部分Protein Metrics合作发表的Poster和Presentation,如果您对此感兴趣也欢迎您参加相应的环节以及到我们的展台咨询! MP051—Monitoring the Aggregation-Induced Conformational Conversion of α-SynucleinProtein by Fast Photochemical Oxidation of Proteins (FPOP) MP300—IntegratedSoftware Platform for Analyzing Hydrogen-Deuterium Exchange and Oxidative Footprinting Data for Solvent Accessibility MP415—An Automated Data Analysis Workflow for Intact and Sub-Unit Mass Analysis of Protein Reagents Using Different Mass …

Don’t Miss Our 2019 Shanghai Workshop, June 18! Protein Metrics致力于为客户提供更好的生物药物的LC-MS数据分析软件平台。目前,针对生物药物的常用表征流程,Byos一站式分析平台已经能够无缝衔接您药物开发不同阶段的分析表征的各种需求,包括蛋白分子量分析,肽图分析,二硫键分析,序列变异分析,MAM,糖链分析等等。同时,在不断完善现有流程的基础上,我们也持续的对软件进行更新和升级,以提供更好的流程和功能。 参加本次研讨会, 您可以获悉国外最新的生物药物表征发展趋势以及Protein Metrics最新的软件功能进展; 您可以与来自Protein Metrics总部的应用专家现场交流如何展示数据,提高分析的效率; 您可以与Protein Metrics的客户现场沟通,共同交流对您最有意义的功能的和需求! A Day In Shanghai with Protein Metrics 欢迎扫描下方二维码或点击阅读原文报名参加!

Byos增加MAM功能以来,不断收获新的成果,来自PMI最新的消息: Protein Metrics Receives FDA RFQ Award for Multi-Attribute Method (MAM) Software “We are thrilled thatthe FDA has chosen to investigate MAM approaches with Protein Metricssoftware,” says Eric Carlson, Ph.D., President and CEO. “While we recognizethat this award does not constitute an endorsement, we are extremely pleased that the FDA recognized …
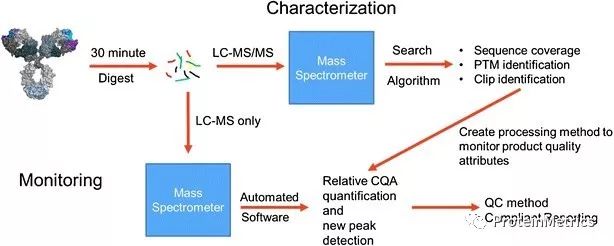
近年来,关于LC-MS在生物药物理化性质表征的话题受到越来越广泛的关注,而生物制药的企业和仪器厂商也都在不断的完善LC-MS方法用于生物药物的研发,放行,稳定性测试等。理化性质表征的多属性方法(Multiple-Attribute Method, MAM),能够解决生物药物分析面对的挑战,减少QC所需的分析测试的实验数量,对于增加生物制药企业效率具有重要的意义。基于LC-MS的MAM在生物药物中的表征方案和流程成为大家密切关注的MAM方向之一,利用新的MAM流程完成所需的分析能够使制药行业加速研究,顺利实现不同时期的方法转换。 目前传统QC肽图定量的工作也逐步从单一的UV分析开始转向利用基于质谱XIC的分析,从而获得准确度更高,专一性更好的结果。肽图分析可以结合MAM同时进行靶向、大量的药物关键质量属性的分析,比如进行序列变异的监测,位点修饰的定量等等。MAM还可以在数据处理时增加New Peak Detection功能,进行比对性研究的时候能够快速的对未知峰进行分析。 MAM Workflow MAM的特异性和定量能力在整个生物制药研发生产过程提供对CQA的控制,以及合并和删除信息量少且冗余的测试方法,对企业来说MAM可以简化生命周期管理活动,从而在维持单一方法的情况下提供监管机构所需要的多种信息。 尽管越来越多的人开始多MAM方法感兴趣,但是,如何去建立一个最佳的MAM方法在您的实验室,您是否需要行业的专家给您提供指导? 本次PMI资深应用团携手MedImmune的专家在十二月六号将给大家带来MAM方法开发的讲座。想参加MAM的Webinar欢迎点击左下角“阅读原文”报名! 参加本次讲座,您将得到如下的信息: 如何在您的实验室建立MAM; 如何同时监测多种分子的属性; 如何在整个流程中管理在MAM分析中的挑战:样品制备,色谱质谱数据采集; 如何应对MAM数据分析和报告的难题。 Reference: Rogers, R.S., Abernathy, M., Richardson, D.D. et al. AAPS J(2018) 20: 7.

PMI以客户的成功为己任,不断更新软件并提供给客户更加强大的工具,Byosv3.2十月份正式上线啦,您是否已经开始下载使用了呢? 新版本都有哪些变化呢?小编带您快速划重点——Byos MAM Workflow! MAM( Multi-Attribute Methods)是生物制药质量控制方法之一。生物制药产品开发过程中,监控治疗药物分子的关键质量属性对于药品的安全性和有效性来说至关重要,目前,生物制品的产品质量属性检测利用一系列的UV,LC,CE等方法,质谱技术作为一次分析同时检测大量产品质量属性的手段。基于质谱的MAM手段可以快速的监测药品的多种不同的肽段,修饰等信息,对于药品的研发和产品放行检测来说都是十分重要的。 Byos新版本增加的四个MAM Workflow,通过LC-MS/MS数据的分析,针对特定峰的信息检测,定量,可以一次性给出多种用户关注的PQA,对于生物药品的研发和质量控制来说是非常便利的方案,解决用户在MAM数据分析和报告中的困难。 – MAM Reference Characterization 是基于LC-MS或MS/MS数据进行目标质谱峰的定性定量; – MAM Chromatogram Annotation (in-silico) 是对LC-MS质谱信息进行目标的定性定量; – MAM Chromatogram Annotation (LC-MS/MS) 是基于LC-MS/MS的结果对目标峰进行注释; – MAM New Peak Detection是基于LC-MSor MS/MS对新的峰进行定性和注释。 当然除了MAM Workflow之外,Byos v3.2 还有其他的功能更新跟升级,以更加符合用户需求,包括Oxidative Footprinting和System Suitability两个新的Workflow,以及Intact Mass计算,峰提取,峰对齐,报告等功能上的更新。 – System Suitability 帮助您验证LC-MS/MS system评价和和验证; – Oxidative Footprinting …