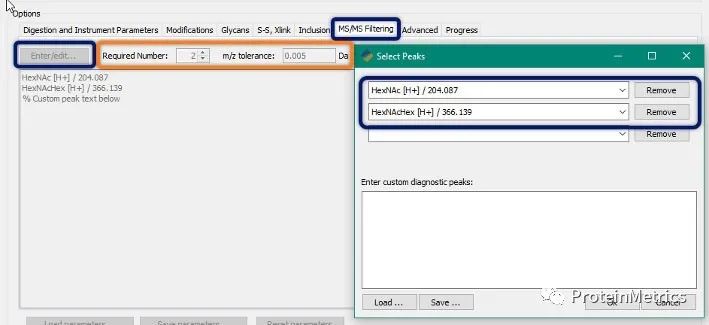
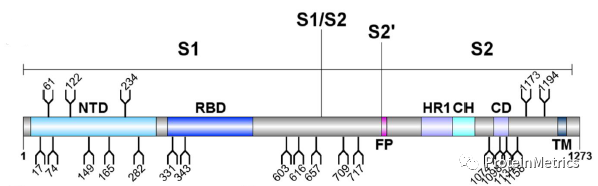
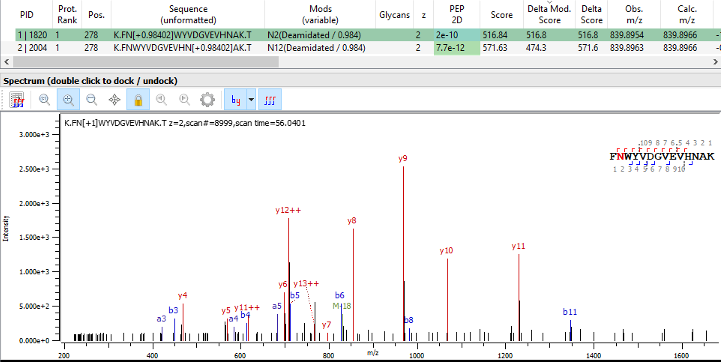
点击上方蓝字关注我们 Don’tMiss Our 2020 Online Workshop, Dec 17! Protein Metrics致力于为客户提供更好的生物药物的LC-MS数据分析软件平台。 目前,针对生物药物表征流程,Byos一站式分析平台已经能够无缝衔接您分析表征的各种需求:蛋白分子量分析,肽图分析,二硫键分析,序列变异分析,MAM,糖链分析,蛋白高级结构分析,质谱软件的网络版部署等等。 本次研讨会,ProteinMetrics 邀请了国内外一流的生物制药领域的专家与大家交流生物制药表征的经验,您可以获悉国内外最新的生物药物表征发展趋势以及ProteinMetrics最新的软件功能进展,共同交流对您最有意义的表征方案和功能! 9:30 AM Log-in Online meeting 9:45 AM Welcome and Introduction 10:00AM Lei Wang, Ph.D., Takeda Lei是武田制药的的首席科学家。他获得了中科院化学所的分析化学博士。在加入制药行业之前,他曾在麻省理工学院进行博士后研究,并在康奈尔医学院担任讲师。Lei在生物质谱技术(尤其是LC-MS)进行生物标志物开发和蛋白表征方面拥有超过10年的经验。在武田制药,他的工作重点是治疗性蛋白质的高级表征,可开发性评估,杂质鉴定和高通量分析等,以确保产品质量,安全性和功效。目前,Lei是美国药典MAM专家小组成员。 Topic: Specificand confident Glycation Site mapping in Therapeutic Proteins Using AutomatedData Processing 10:30AM 汪彤丹,博士,药明生物 上海药明生物技术有限公司分析科学部副主任,药明生物质谱卓越中心主要负责人。博士毕业于中国科学院上海有机化学研究所,曾在上海交通大学医学院从事多年的基于质谱的蛋白质组学研究工作。自2016年始加入药明生物,主要从事蛋白质质谱表征,包括方法开发,结构表征比较性研究,肽谱图放行方法开发,评估,确证等。带领团队参与在研生物药的质谱表征和临床各期工艺研发,包括单抗、融合蛋白、双特异性抗体、ADC、酶、疫苗等。 演讲题目:基于质谱的生物药结构表征策略与案例分享 11:00AM Q&A 点击阅读原文注册参会,会后将从注册名单抽取幸运观众,也欢迎大家扫码加入PMI Workshop微信群,与我们交流生物制药LC-MS数据分析,会议期间微信群不定时会发送红包雨。 ✎ 联 …
诚邀您参加Protein Metrics 2020 Online Workshop Read More »