



第二期
糖肽的RPLC
-MS/MS行为
Protein Metrics 高准
RPLC行为
离子活化
糖肽裂解规律
分子重排
错误匹配来源
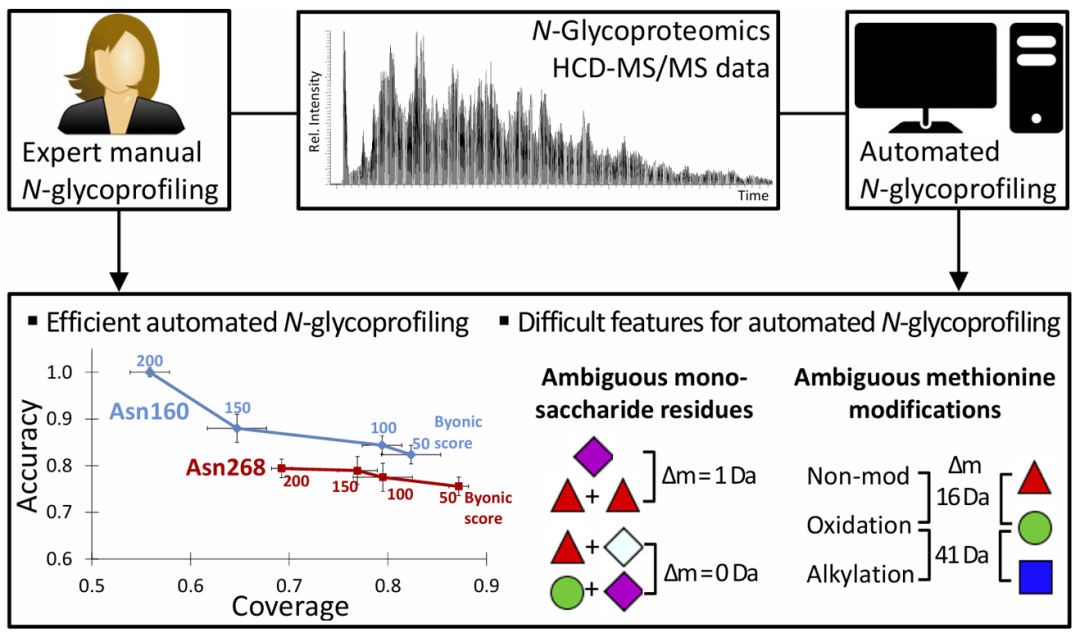
Byos中糖肽结果验证
本文简要介绍糖肽的RPLC-MS/MS行为,主要包括色谱保留,离子活化,质谱裂解规律,分子重排和错误匹配来源等,帮助分析学家快速读懂糖肽LC-MS/MS图谱。
01
RPLC行为
一个糖肽二级谱往往可以匹配多个不同的聚糖和肽段组合,尤其是当质谱数据质量较差(缺少特征诊断离子或者前体离子测定不正确),保留时间就为糖肽鉴定提供非常重要的互补信息。
糖肽疏水性越强,在RPLC上的保留时间越长。通常糖链上引入中性单糖,如Hex和HexNAc,会增加糖肽亲水性,导致保留减弱。不改变电荷的翻译后修饰,对保留时间影响不大,蛋氨酸氧化除外。有研究发现,如果聚糖链上存在 NeuAc 或磷酸基团,则会导致保留时间增加,因为带负电荷的 NeuAc 和磷酸基团会抵消糖肽质子的正电荷,从而与反相色谱柱更紧密地结合。

02
离子活化
目前糖肽常用的解离方法有CID,HCD和ETD等,根据需求选择合适的碎裂方法。
CID
由于解离能低和1/3 cut-off,离子阱CID主要产生Y离子,伴有少量的B离子和b/y离子。该技术可识别结合的聚糖,但无法给出准确的糖基化位点和肽序列,可用于简单样品中的糖肽分析。
HCD
HCD可以生成丰富的B/Y互补离子鉴定糖部分;随着碰撞能量增加,b/y系列离子会增多,可鉴定肽部分。研究表明,HCD–MS/MS以20-30-40%的能量可生成信息量最多的聚糖和肽相关碎片离子。
ETD
ETD主要产生c/z离子,可用于鉴定糖基化位点和肽序列。由于ETD效率较低,通常与HCD或者CID结合使用,即EThcD。随着离子触发技术发展,避免了ETD在非糖肽解离上浪费的反应时间。
03
糖肽裂解规律
在糖质谱命名法中,A/B/C是非还原端离子,X/Y/Z是还原端离子。
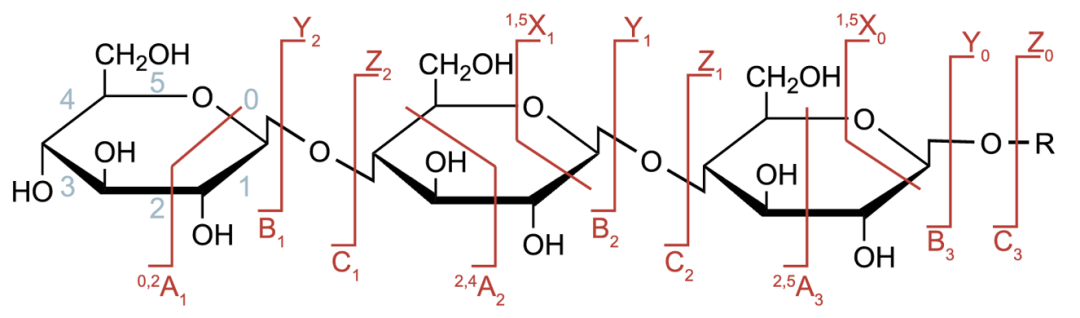
B离子是糖肽重要的诊断离子,可用于糖型判断,触发ETD,二级谱过滤等。常见的B离子,如下图所示:
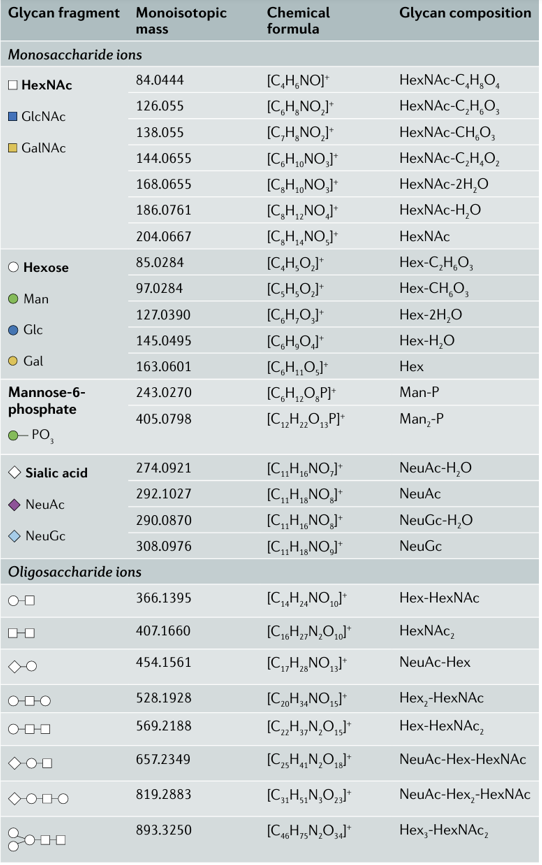
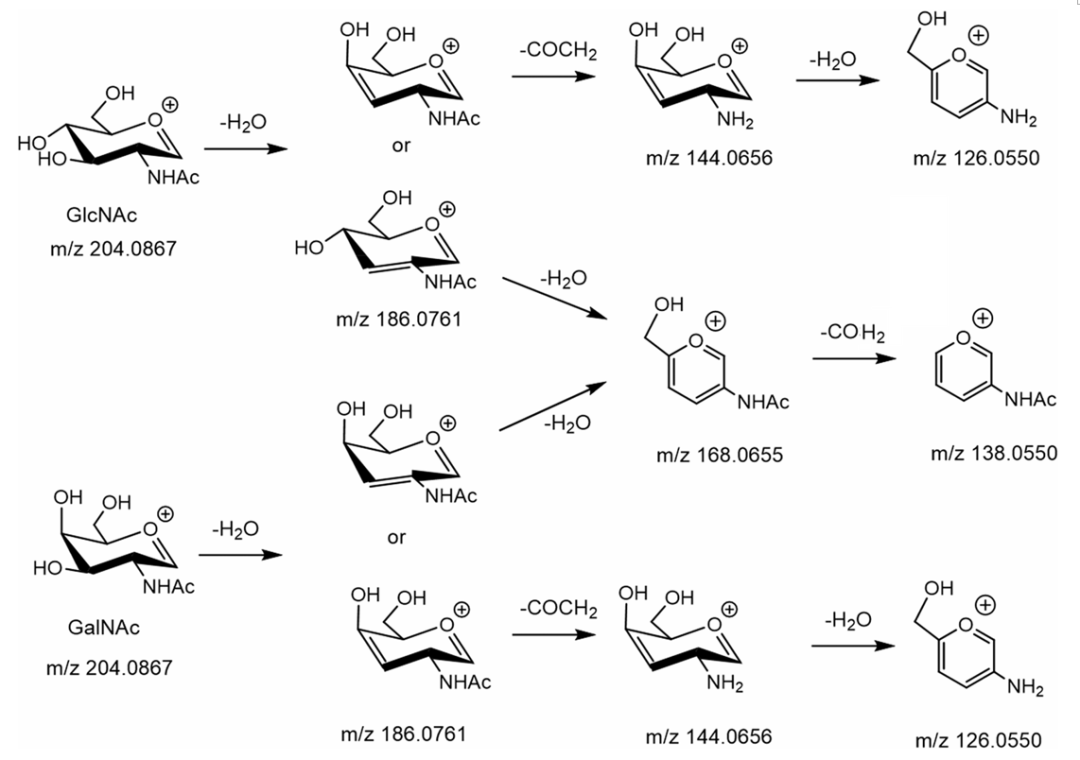
B离子在碰撞池中会发生进一步碎裂,通常会丢失H2O,CH2=C=O,HCOH等,我们以HexNAc为例,展示单糖的质谱裂解规律,如下图所示:
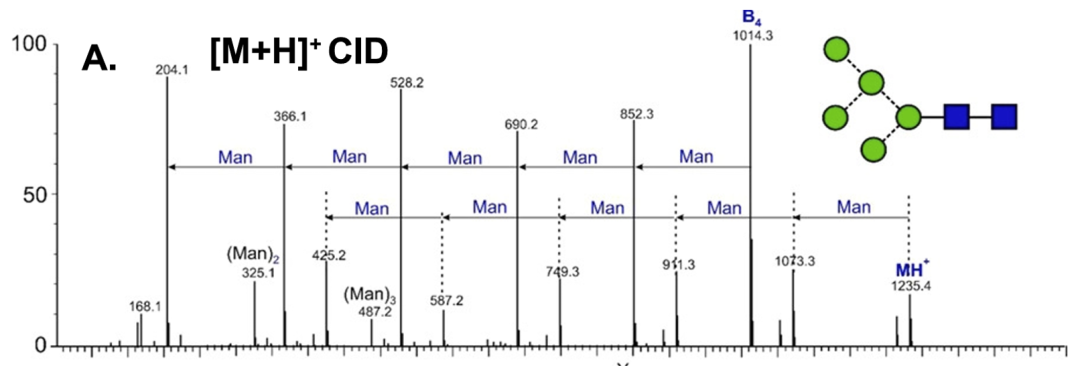
在糖肽结构中,往往是糖的还原端与肽段直接相连,因此Y离子由Peptide+Glycan两部分组成。HCD二级谱中Y离子连续的单糖丢失,可用于糖组成和拓扑结构鉴定。
04
分子重排
质子化糖肽在碰撞池中容易发生分子重排,如单糖迁移(Fuc和其他单糖)和内部残基丢失(可以是单个单糖,也可以是多个单糖,通常以中性碎片形式离去),进一步复杂化糖肽二级谱,导致糖链拓扑结构鉴定错误。全甲基化和乙酰化的糖肽也容易发生分子重排。
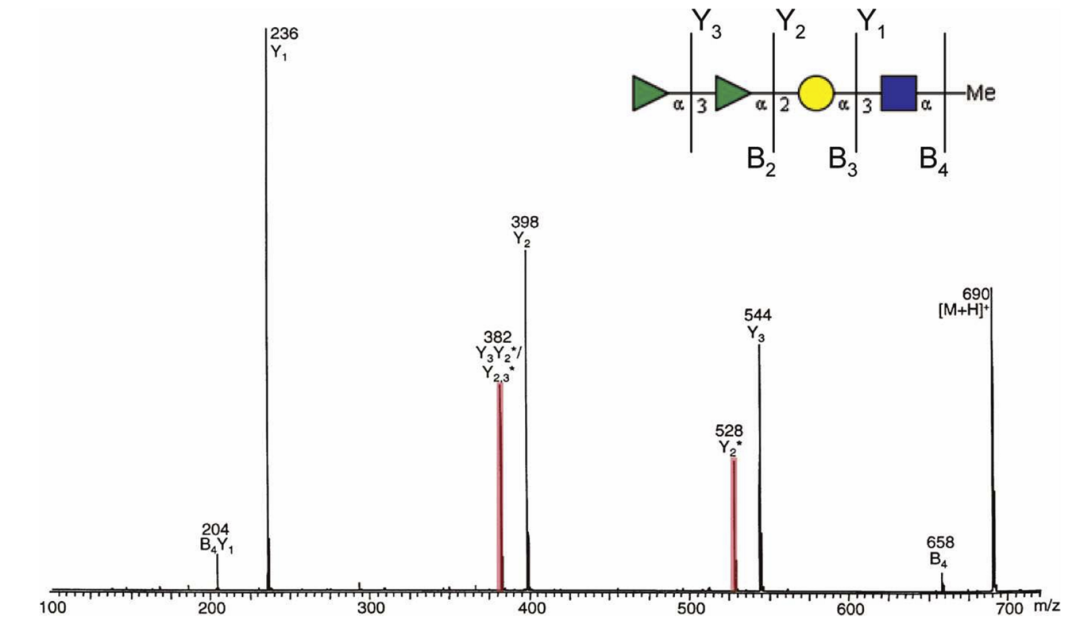
Y2*表示还原端第二个糖丢失,Y3Y2*表示还原端第二个和第三个糖丢失。
为了消除重排干扰,可通过去质子化糖肽或者糖肽碱金属加合物来分析糖链结构。研究表明,去质子化和碱金属加合物糖肽在质谱中不会发生重排反应。
05
错误匹配来源
常见的糖肽匹配错误来源有两个:单同位素峰鉴定错误(如NeuAc与2Fuc质量差为1)和肽段修饰分子量与单糖分子量之差相等(如氧化+NeuAc与NeuGc质量相等)。
06
Byos中糖肽结果验证
Byos软件提供多个维度自动化的糖肽结果验证指标,如MS1 Correlation,Score,PEP2D等,释放手动解谱压力,使大规模糖肽组分析成为可能。
MS1 Correlation评估实际观测糖肽同位素峰分布与理论同位素峰分布的相关性,降低单同位素峰鉴定错误导致的糖肽错配概率。Byos 3.7及后续版本引入了惩罚性打分,如NeuAc不含有274,NeuGc不含有290等,最小化修饰肽造成糖肽匹配错误。
通常Score与PEP2D搭配使用,Score > 200,PEP2D < 0.001,基本可以保证糖肽鉴定都是正确的。
结语
本文是糖蛋白系列文章第二篇,主要介绍糖肽的RPLC-MS/MS行为,帮助您快速解析糖肽二级谱。后面还有更多精彩内容,敬请期待吧。
参考文献:
向上滑动阅览
[1] Bagdonaite, Ieva, et al. “Glycoproteomics.” Nature Reviews Methods Primers 2.1 (2022): 48.
[2] Cao, Weiqian, et al. “Recent advances in software tools for more generic and precise intact glycopeptide analysis.” Molecular & Cellular Proteomics 20 (2021).
[3] Wang, Benlian, et al. “Reliable determination of site-specific in vivo protein N-glycosylation based on collision-induced MS/MS and chromatographic retention time.” Journal of The American Society for Mass Spectrometry 25.5 (2014): 729-741.
[4] Ang, Evelyn, et al. “Retention time prediction for glycopeptides in reversed-phase chromatography for glycoproteomic applications.” Analytical chemistry 91.21 (2019): 13360-13366.
[5] Klein, Joshua, and Joseph Zaia. “Relative retention time estimation improves N-glycopeptide identifications by LC–MS/MS.” Journal of proteome research 19.5 (2020): 2113-2121.
[6] Wuhrer, Manfred, André M. Deelder, and Yuri EM Van Der Burgt. “Mass spectrometric glycan rearrangements.” Mass spectrometry reviews 30.4 (2011): 664-680.
[7] Kuo, Chu‐Wei, et al. “An N‐glycopeptide MS/MS data analysis workflow leveraging two complementary glycoproteomic software tools for more confident identification and assignments.” Proteomics 23.20 (2023): 2300143.
[8] Lee, Ling Y., et al. “Toward automated N-glycopeptide identification in glycoproteomics.” Journal of proteome research 15.10 (2016): 3904-3915.
[9] Grabarics, Márkó, et al. “Mass spectrometry-based techniques to elucidate the sugar code.” Chemical Reviews 122.8 (2021): 7840-7908.
关于Protein Metrics
Protein Metrics LLC是一家全球领先的质谱数据解析软件供应商,公司总部位于美国波士顿。我们为科研和企业用户提供高效准确的一站式质谱数据解析方案,帮助用户发现、解决问题。Protein Metrics在全球范围内提供销售和支持,目前已为超过200个企业和300个科研单位提供服务。
联系我们邀约演示:
王蕾 13482181958