




序列变异体(SVA)
分析应用指南
Protein Metrics – Customer Success Team
简介
几乎所有治疗蛋白质数据集都会包括质谱数据,其中均包括肽图分析,在一定浓度下,这些肽与数据库中对应的肽段序列对比,可能会存在实际与理论的序列不一致的情况。序列变异体可能是由宿主细胞系中的突变或错误翻译、生物反应器进料问题或正常生物噪音引起的。搜索序列变异体特别具有挑战性,因为在此过程中可能存在大量的氨基酸取代,并且许多质量增量与翻译后修饰和样品制备伪影导致的质量增量完全相同或大致相同。在本应用说明中,我们展示了如何搜索和量化生物治疗药物中的序列变异体。
序列变异体分析旨在识别和量化所有变异肽,直至所占百分比极小的“野生型”(天然或预期序列)对应物。由于治疗蛋白质的蛋白质数据库非常小,通常只有产品加上胰蛋白酶或其他消化酶和诱饵,因此 Byonic 运行时间并不是问题所在。我们也可以在几分钟内搜索所有可能的单-AA-置换肽。其挑战在于区分相对罕见的真实序列变异体和误报结果。
本技术说明介绍了如何使用 ByosTM(传统 ByonicTM 和 Byologic®)确定 MS/MS 数据中的 SV,其中还总结了最佳序列变异体 Byonic 搜索的最佳实践注意事项。请注意,搜索的质量在很大程度上取决于质谱分析的质量,因此本文中还会提到 MS 分析技巧。
' fill='%23FFFFFF'%3E%3Crect x='249' y='126' width='1' height='1'%3E%3C/rect%3E%3C/g%3E%3C/g%3E%3C/svg%3E)
摘要
执行 Byos SVA 工作流程
分析技巧
o 酶切设置
o 修饰、聚糖和输入设置
配置工作流程
检查结果
o 使用 Byos 消除明显的假阳性结果
o 检查 MS1 和 XIC
o 检查 MS2
o 其他提示
生成报告
执行 Byos SVA 工作流程
单击 SVA 启动工作流程

拖放原始数据文件
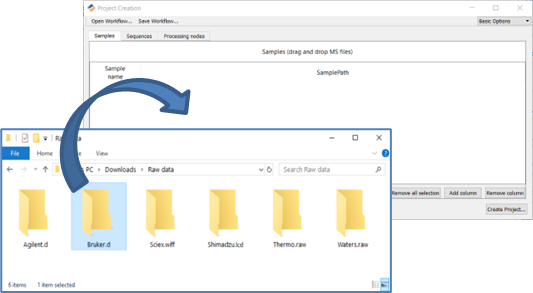
拖放 FASTA 文件
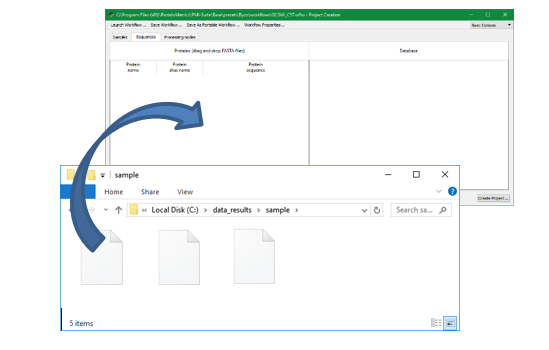
创建一个项目
分析技巧
烷基化剂的选择
许多氨基酸置换具有与常见烷化剂完全相同的质量差。我们建议根据所需的分析选择一种:
o C13 标记的碘乙酸 (+59 Da) 与可能的氨基酸置换没有重叠,但获得成本更高。
o 碘乙酸 (+58) 相较于碘乙酰胺生成的过烷基化肽相对更少,并且与所有可能的氨基酸置换列表的质量增量重叠更少。
o 然而,如果仅考虑导致序列变异体的单碱基置换,而不是整个列表,则碘乙酰胺 (+57) 可能是所需的试剂,因为在这个相对较短的列表中不存在冲突的质量。
使用高分辨率质谱仪
使用高分辨率 MS1。理想情况下,还应使用高分辨率 MS2。确保仪器经过适当校准并以接近峰值的性能运行。
考虑使用 PreviewTM
首先使用 Protein Metrics 的 PreviewTM 或 Byonic Wildcard SearchTM 识别样品问题。如果 Preview 表明烷基化或消化不理想,请考虑重新运行分析。样品制备伪影可能会产生许多误报结果.
考虑将序列变异体搜索分成多个阶段
或者,开始一项研究时,重点列出由于核糖体摆动(通常低浓度)和/或其他可疑变异体引起的预期置换,然后扩展到所有潜在氨基酸置换的完整列表。
配置工作流程
MS/MS 搜索 – 消化设置
使用完全特异性(fully specific)的酶切设置,产生零个或一个错误切割。如此一来,结果将更加准确,误报更少。一般来说,第一个使用的酶是胰蛋白酶。然而,这可能会错过涉及 R 和 K 的变异体。目前有几种策略可以挑出这些变异体:
o 可能对胰蛋白酶消化样品进行第二次 Byonic 搜索作为半特异性(semi-specific),并将注意力集中在涉及 R 和 K 的假定变异体上。
o 考虑在您的方法中加入 ETD 碎裂模式,来提高肽段的覆盖率,更好的找到变异体。
o 此外,第二种酶(单独处理和分析),如胰凝乳蛋白酶(chymotrypsin)或嗜热菌蛋白酶(thermolysin),对于验证和/或发现其他变异体很有帮助,因此推荐使用。对于具有许多酶切位点的酶,使用“-1”表示漏切的数量。
MS/MS 搜索 – 修饰、聚糖和输入设置
仔细自定义设置您的 Byos SVA MS/MS 搜索可以得到质量更高的 序列变异体分配。默认的 Byos SVA 工作流程已包含许多建议,但其中一些建议主要是针对实验室,因此应进行评估。
o 不要将脱酰胺作用(deamidation)纳入变异体列表 (Asn->Asp)。相反,应将其命名为 rare1 修饰。列出样品中已知存在的所有类型的非变异修饰,其中包括您实验室特有的所有样品制备。除用于 pyro-glu 修饰的 common1 以外,所有修饰和序列变异体均使用 rare1。
o 下表显示了典型的修饰列表。如上所述,一份好的列表应包括所有预期的 PTM 以及任何常见的样品制备伪影和常见的实验室加合物。
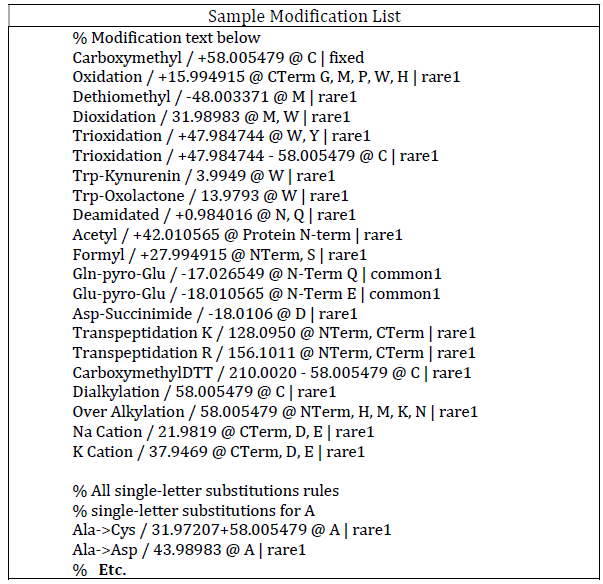
o “Total Common Max”和“Total Rare Max”均应设置为 1。
o 默认 Byos 工作流程中包含所有单个氨基酸变化的集合。Protein Metrics 还提供了包含序列变异体所有可能修饰的其他工作流程。如果手动将自定义序列变异体添加到列表中,请将搜索中的序列变异体设置为 rare1 类型修饰。
o 此外,对于具有 N-糖基化的 mAb,请在“Glycan”选项卡上添加包含 50 个 N-聚糖的列表。
o 在”Spectrum Input Options”中,确保将“Maximum # of precursors per scan”设置为 1。这将减少搜索结果中的误报。
检查结果并创建报告
通过 Byos 进行序列变异体 (SV) 检查的目标是快速识别误报结果、验证真实结果 SV、根据 XIC 比率量化变异体并生成报告。下文提供了最佳实践的分步列表,以最大限度地缩短分析时间并提高结果的置信水平。
使用 Byos 消除明显的误报结果
o 设置预定义的筛选器(search filter)来限制项目范围。删除不是序列变异体或其相应野生型肽的候选肽。另外,设置合理的分数截止点。这取决于数据集,但通常开始分数设置为 Byonic 分数 > 100 比较理想。同时,还需要设置数据相应的最大前体误差。
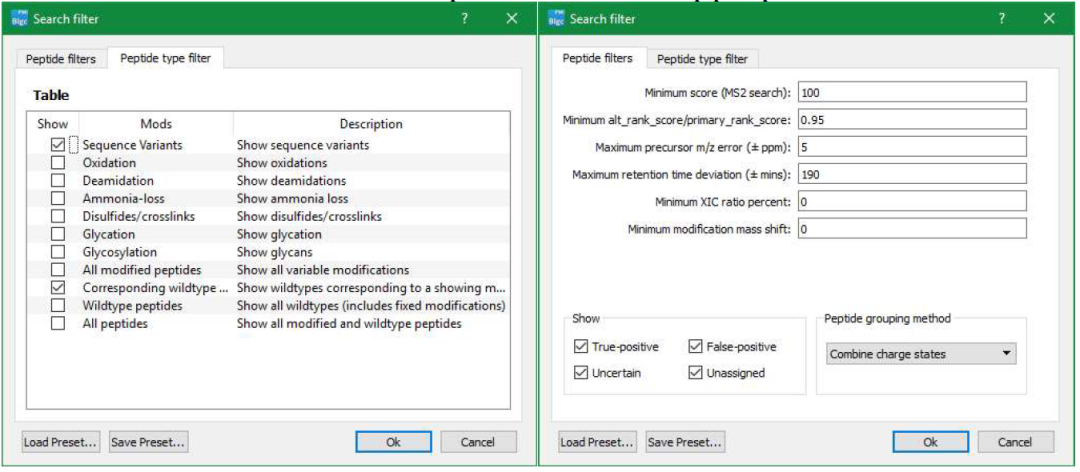
o 如果使用具有完全特异性(fully specific)和半特异性(semi-specific) SV 搜索的参数,请检查是否在每个参数中均识别出了假定 SV。如果不是,则可能是误报。突出显示该行并按 CTRL+F 将其标记为“False-positive”(或双击“Validate”列并选择“False-positive”)。使用 Comment 字段为选择添加任何上下文。
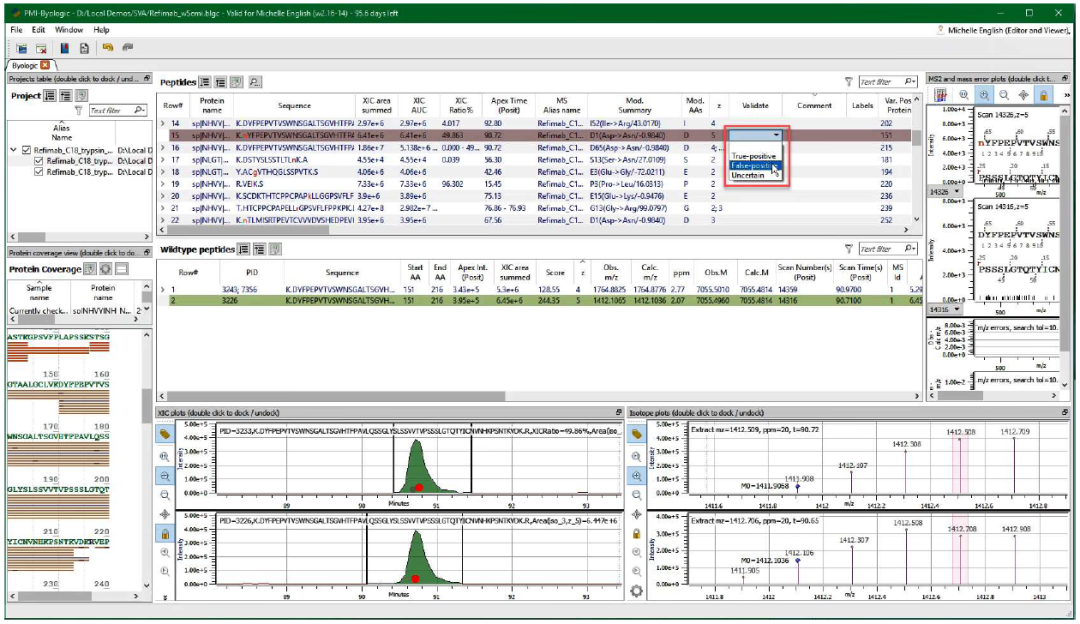
o 所有已识别的 SV 必须具有与其相关的野生型(未修饰)肽。如果没有野生型肽,则标记为误报。可以通过对此类误报进行排序来归类肽表中的假定变异体,并通过首先突出显示该组的所有行并单击“Ctrl F”在一个操作中标记为误报。(涉及酶切位点的 SV 可能是该规则的例外情况。请参阅下文了解这些修饰。)
联系 Protein Metrics 支持团队获取动态筛选器文件,该文件将突出显示没有相应野生型的所有行。“No Wildtype”和许多其他预定义筛选器都可以保存到本地 PC。保存筛选器后,从peptide可视化界面中选择筛选器图标。选择“Presets”、“Import from file”、“Select file”并导航至保存的筛选器。
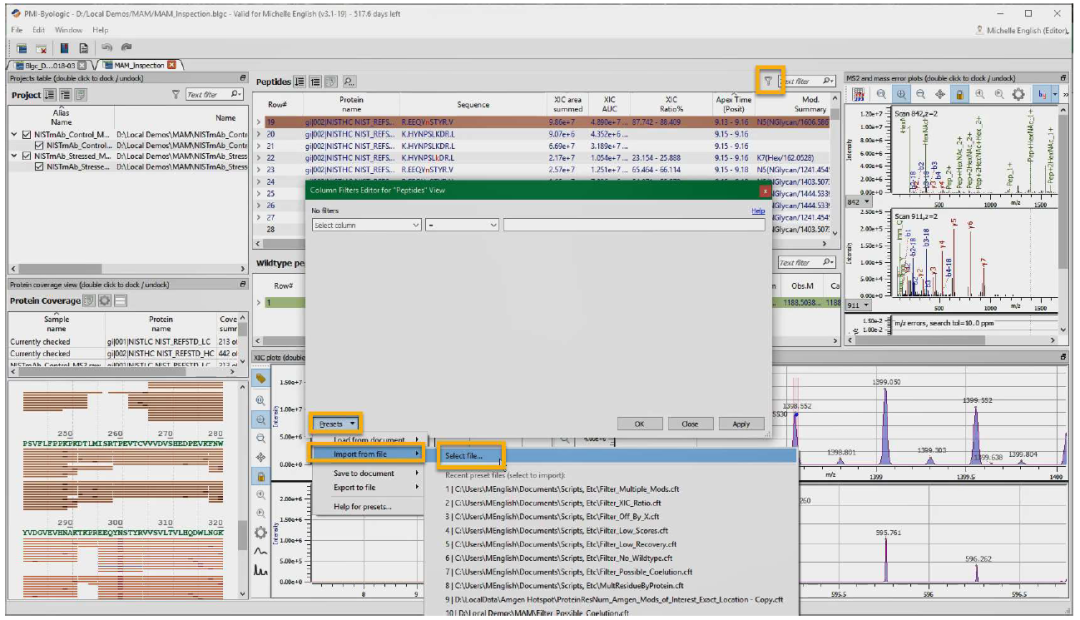
o SV 是否与另一个修饰一起出现,并且仅与该修饰一起出现?标记为误报。还提供双修饰肽的筛选器。
检查 MS1 和 XIC
回答以下一系列问题可以帮助您确定候选肽的真实性。
o 查看 MS1 同位素剖面并找到蓝点。蓝点是在单一同位素峰上吗?如果不是,则标记为误报。通常这些错误也被标记为“Off-by-X”,并且可以使用筛选器突出显示。
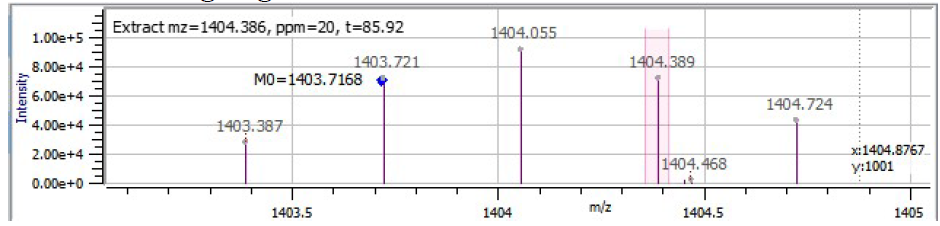
o 电荷分配是否正确?如果不是,质量将被错误计算,并且该行应被标记为误报。
o SV 的保留时间变化是否合理?SV 通常会引起疏水性变化,这一点在肽的保留时间上有所体现。
o 检查肽表中变异体的丰度(XIC 比率)。如果丰度非常低,请考虑将其标记为“Low Abundance”,然后继续。可配置的筛选器也可以帮助完成这项工作。
– 如果残基有多种可能的变异体,请稍后返回这些内容。累积丰度可能很大。
– 确保 XIC 积分窗口大致正确,因为这会影响 XIC 比率。在此阶段,不需要达到理想状态。
o SV 的保留时间变化是否合理?SV 通常会引起疏水性变化,这一点在肽的保留时间上有所体现。
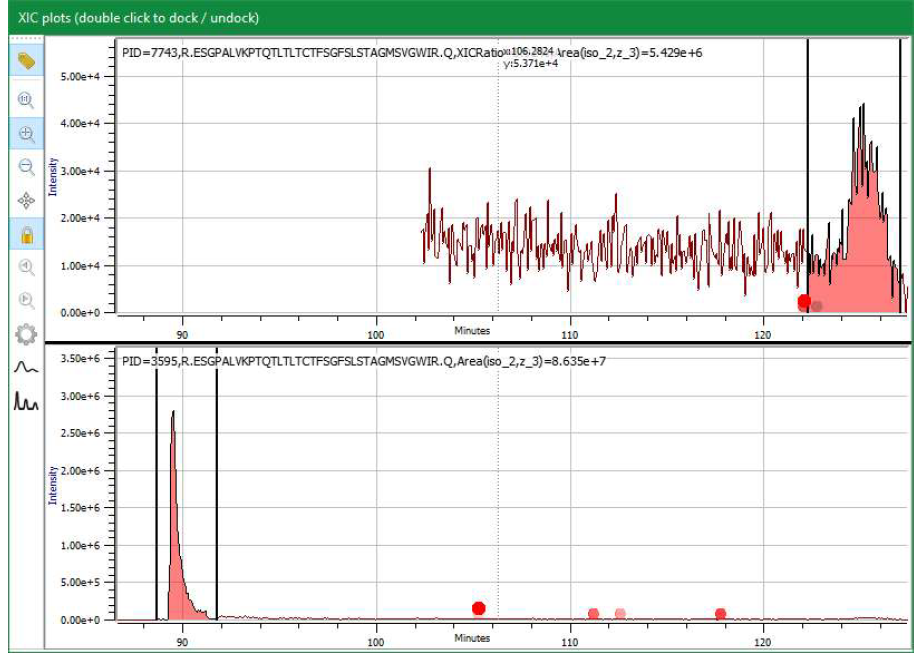
Byos 计算任何序列变异体的预期保留时间,并提供一个字段,显示保留时间中观察到的变化和预测变化之间差异(“Delta RT Observed – Delta RT Predicted”)。如果保留时间的变化不合理,则标记为误报。可配置的筛选器可帮助突出显示这些情况。
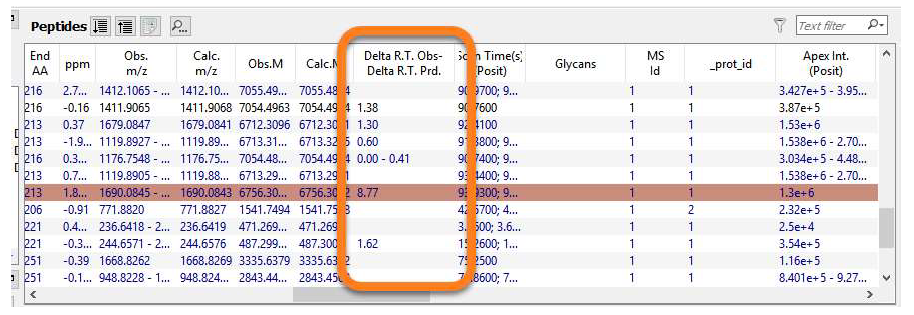
o MSMS 结果(红点)是否与 XIC 相对应?
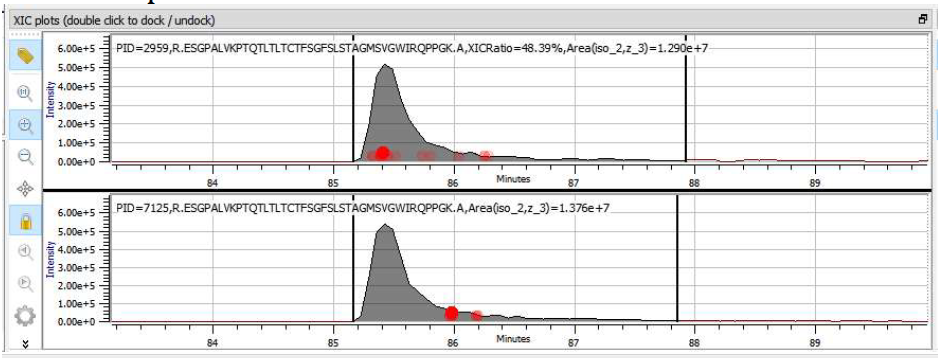
o SV 是否与其野生型共洗脱?与野生型完全共洗脱的蛋白质种类很可能是柱后或源内发生的假阳性。筛选器可突出显示这些内容以便快速识别。
检查 MS2
o 野生型和 SV 中识别出的碎片离子应该相似。
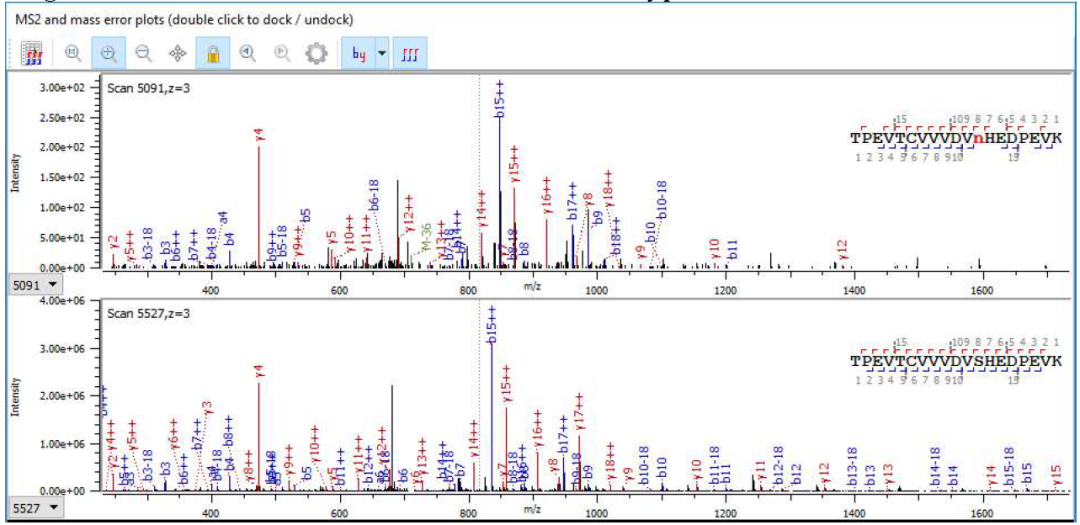
o 碎片离子是否与噪声水平下的 SV 相对应?这可能是误报。
o 是否存在高强度但尚未分配的碎片离子?如果相应的离子分配在野生型中,这仍然可能是 SV 修饰,但位于肽上的不同位置。
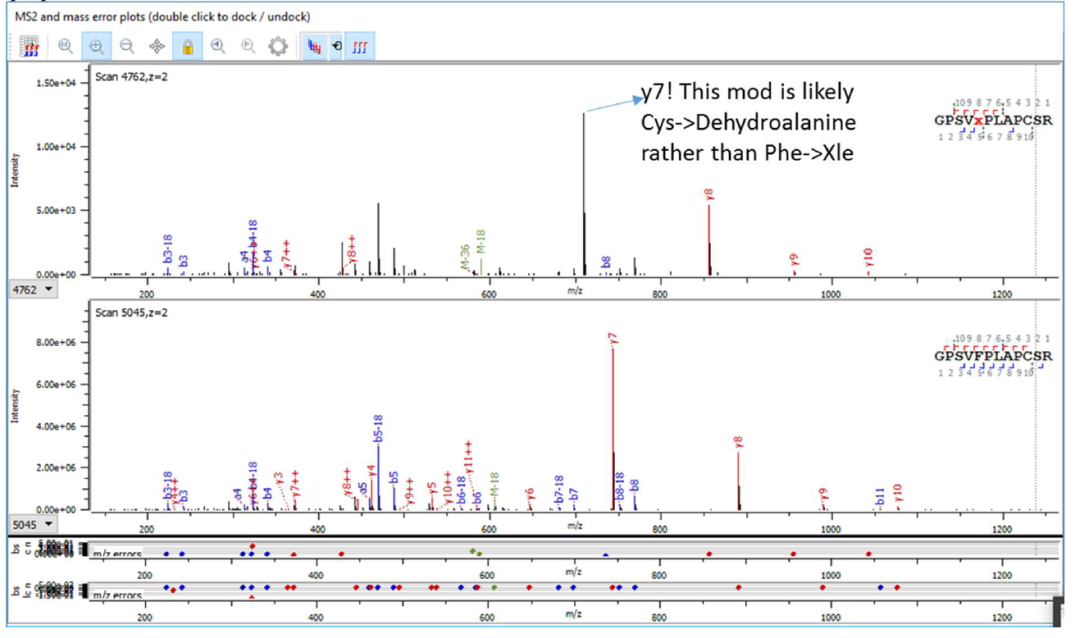
o 离子强度和中和损失是否正确?例如,脯氨酸 y 离子应为高强度,N、Q、K、R 失去氨基 (-17 Da),S、T 和 E 失去水 (-18 Da)。
其他提示
o 在“Show All Peptides”处于激活状态的情况下打开第二次出现的 Byologic 项目方便快速查看包括野生型在内的其他肽形式,而主要注释则集中在序列变异体,或者,使用带有“Ala->”文本的筛选器逐个筛选氨基酸。
o 对于涉及 Lys 和 Arg 的变异体,尽管建议对这些 SV 使用除胰蛋白酶之外的第二种甚至第三种酶,但可以使用完全特异性搜索来查找这些变异体。
– 对于 K/R -> X 的情况,使用 Byos 搜索,其中有 1 个遗漏切割且具有完全特异性。当加载到 Byos 中时,仅将注意力集中在具有 1 个遗漏切割和 K/R -> X 的候选肽上。还可以考虑限制胰蛋白酶的消化时间,从而有意诱导漏切。
– 对于 X -> K/R 的情况,执行半特异性酶切 Byos 搜索,并仅将注意力集中在新出现的肽上,并根据 X -> K/R 进行切割。
o 较大的胰蛋白酶肽通常质谱质量较差,从而导致难以进行 SV 检测。如果可以的话,考虑运行 ETD 以确保肽段的序列覆盖。
摘要
分析完成后,Byos 不仅会打开检查视图,还会在 Byos 窗口中打开相应的报告作为选项卡。如果报告选项卡曾经关闭,可以通过单击菜单栏正下方的报告选项卡或选择“FileàExportàGenerate configurable pivot summary…”重新打开
默认报告配置包括蛋白质覆盖率、包含 SVA 信息及其丰度的详细数据透视表,以及每个样品的每个氨基酸的平均修饰百分比的表格。但是,可以通过选择“FileàPresetsàReport PresetsàBlgc_SVA.rptc”生成 SVA 特定报告。报告示例具体如下所示。
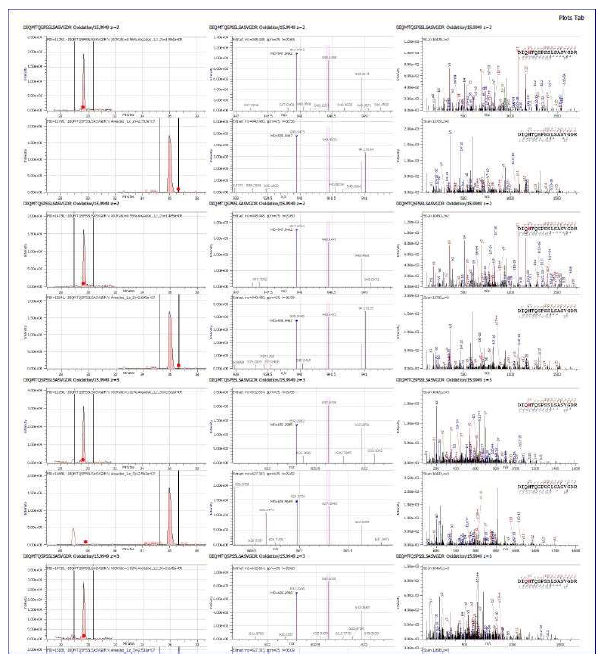
若要将质谱和 XIC 图像纳入报告,请单击“TabsàAdd Plots”。此步骤可能需要几分钟,具体取决于分析中包含的肽段数量。
在导出前隐藏配置字段。
单击“File->ExportàExport to PDF…”生成包含所有选项卡的单独 PDF 文件。
有关如何自定义此报告的更多信息,请参阅 https://www.proteinmetrics.com/resources/#videos-tutorials 上提供的相关视频,或通过 support@proteinmetrics.com 联系Customer Success Team。
关于Protein Metrics
Protein Metrics LLC是一家全球领先的质谱数据解析软件供应商,公司总部位于美国加州。我们为科研和企业用户提供高效准确的一站式质谱数据解析方案,帮助用户发现、解决问题。PMI 在全球范围内提供销售和支持,目前已为超过150个企业和300个科研单位提供服务。
联系我们邀约演示:
王蕾 13482181958

