


 本期“ Protein Metrics Insights”的重点是Intact Mass。我们将讨论如何对isotopically resolved 谱图进行去卷积分析,然后确定蛋白质的单同位素质量,本期的新功能将介绍Intact Mass的完整分子量重建功能。
本期“ Protein Metrics Insights”的重点是Intact Mass。我们将讨论如何对isotopically resolved 谱图进行去卷积分析,然后确定蛋白质的单同位素质量,本期的新功能将介绍Intact Mass的完整分子量重建功能。
同位素分辨的数据去卷积
通常Intact Mass进行去卷积的参数是得到蛋白的平均分子量信息的。如果您的实验室具有同位素分辨的数据并需要去卷积分析,Intact Mass中需要提供特殊的参数,通过参数的设置允许软件在去卷积之后保留原始谱图中的同位素信息。参数的设置也很简单,只需要降低m / z和Mass smoothing 和Spacing等参数的值。以下是一套比较通用的参数:

对于极高分辨率的数据,用户可以进一步降低上述参数以避免数据因Smoothing造成的数据丢失。
Intact Mass进行同位素分辨的数据去卷积分析时,不要忘记在Advance Command中输入如下的命令:
[Intact]
MaxMonoisotope = 30000
使用此Advance Command后Intact Mass将计算单同位素分子量并将它们放置在Masses表中的“Mass monoisotope”列中。Intact Mass将使用不同的算法去计算所有提取到的峰所对应的单同位素分子量。对于低于30000分子量(MaxMonoisotope参数的值)的信号,Intact Mass将假设质量峰是同位素分辨的,并且将计算的同位素峰与去卷积后峰匹配。对于大于30000的质量,Intact Mass将假设质量峰不是同位素分辨的,它将直接从去卷积的平均分子量中提取校正因子。校正因子是平均分子量质量与平均单一同位素质量之间的差异。
另外一点需要注意的是,MS1彩色点设置为0.6 /电荷内的最高点,这可以防止来自平均质量数的有色点落在同位素分辨的m / z峰之间。
完整分子量重建
分析化学中的一个挑战是如何在系统的研究中能够以正交的方法进行数据结果的对比,从而达到对结论的多维确证。2019年6月发布的Byos/ Intact Mass中,我们引入了一种从肽图谱数据重建蛋白质完整分子量的方法,我们可以通过肽图层面对蛋白酶解之后的肽段和修饰的定量信息,重建理论上的完整分子量信息。在完整蛋白水平上的测得的谱图可以与理论重建的完整分子量谱图进行叠加,通过完整分子量重建的功能,我们提供了一种比较肽图数据与完整分子量数据的方法,这样就可以使用两个谱图之间的差异来比较不同方法之间结果的相关性。
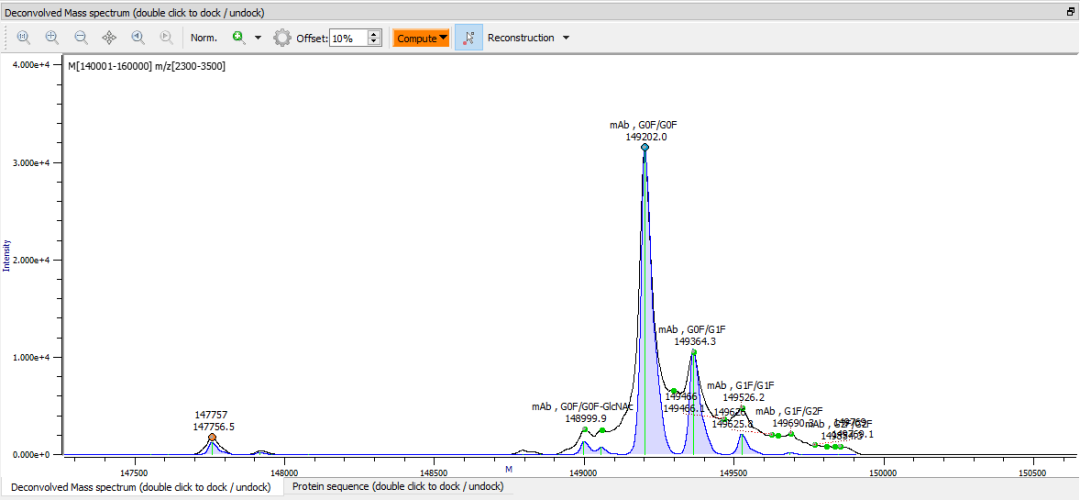 如果您目前正在使用Intact Mass,可以下载beta完整分子量重建演示包可在我们网站的“当前版本”部分中找到。
如果您目前正在使用Intact Mass,可以下载beta完整分子量重建演示包可在我们网站的“当前版本”部分中找到。
Upcoming Webinar:
Automation of Glycan and Peptide Mapping-Leveraging Software to Improve Efficiency and Quality
September 4 Thomas Wesley Powers, Principal Scientist, Pfizer
PTM Analysis-How to Marry Speed and Confidence in R&D
September 17 Jon Reed, Mass Spectrometry Research Scientist, Boehringer Ingelheim
如您有更多需求,请访问我们的网站或联系我们以了解有关Byos工作流程的更多信息。